January 24, 2026
medical device QMSISO 13485 complianceFDA QSRmedtech quality systems
Your Guide to a Medical Device Quality Management System
Master compliance with our complete guide to a medical device quality management system. Learn how to navigate ISO 13485, FDA QSR, and core processes.
19 min readAI Gap Analysis

A medical device quality management system, or QMS, is your company's rulebook for making safe and effective products. It’s the entire framework of processes, procedures, and responsibilities you follow to ensure every single device, from concept to post-market monitoring, consistently meets both customer and regulatory requirements. Think of it as the operational blueprint that guides everything you do.
Deconstructing the Medical Device QMS
A medical device quality management system isn’t just a dusty binder of documents sitting on a shelf. It's the central nervous system of your entire operation—a living, breathing system that connects every department and process with the common goal of producing devices that work as intended and don't harm patients. Without it, a company is just reacting to problems, not preventing them.
To really get it, picture an airline's operational command center. That hub doesn’t just track flight paths. It coordinates everything: meticulous maintenance schedules, rigorous pilot training, air traffic control communications, and emergency protocols. Every move is deliberately planned and executed to guarantee passenger safety.
A medical device QMS works the same way, integrating all critical functions into one cohesive system. This approach ensures quality isn't just checked at the end; it's built into the product from the very first sketch.
Core Purpose and Benefits
At its heart, a QMS is about creating a culture of proactive quality assurance instead of reactive problem-solving. It gives you a structured way to manage every stage of your device's lifecycle. Being proactive is non-negotiable for a few critical reasons:
- Ensuring Patient Safety: The number one goal is to prevent harm. A QMS does this by systematically identifying and neutralizing risks long before a device ever reaches a patient.
- Achieving Regulatory Approval: Agencies like the FDA and other global authorities won’t even consider granting market access without proof of a robust, functioning QMS.
- Building a Sustainable Business: A solid QMS cuts down on expensive errors and recalls, makes your operations more efficient, and builds essential trust with clinicians and patients.
A well-implemented QMS breaks quality out of its silo and makes it everyone's job. It creates a predictable, repeatable, and verifiable system that can withstand the intense scrutiny of regulatory audits and, most importantly, protect the people who rely on your devices.
The market's explosive growth really highlights how vital this is. In 2023, the global Medical Device Quality Management System market was valued at around USD 2 billion and is expected to climb as high as USD 6 billion by 2033. This surge is fueled by increasingly strict regulations that demand comprehensive quality controls from initial design all the way through post-market activities. You can discover more about the market drivers for medical device QMS to see why investing in a quality system is no longer optional—it's a core business imperative.
Decoding the Regulatory Map: Global Standards and Regulations
A medical device quality management system isn't built in isolation. It has to be grounded in a solid framework of global standards and regulations. The best way to think about these rules isn't as bureaucratic red tape, but as a shared blueprint for safety and effectiveness that’s understood worldwide.
For years, anyone in the medical device space has had to navigate two major compliance frameworks. Getting a handle on how they relate to each other is the first real step in building a quality system that will pass muster with auditors and regulators.
The Two Pillars of QMS Compliance
The main global standard everyone works from is ISO 13485. It lays out the essential requirements for a QMS, ensuring that a company can consistently deliver medical devices and related services that meet both customer needs and all applicable regulatory demands.
Meanwhile, in the United States, the playbook has always been the FDA's Quality System Regulation (QSR), found in 21 CFR Part 820. While ISO 13485 and the QSR aimed for the same goal—safe and effective devices—they used slightly different language and had different areas of emphasis. This often meant companies selling in both the U.S. and abroad were stuck doing the same work twice, just to satisfy both sets of rules.
Now, things are changing for the better. ISO 13485:2016 has become the undisputed global benchmark, with nearly 49% of life sciences firms adopting it. The real game-changer, though, is the FDA's decision to finally replace the old QSR with the new Quality Management System Regulation (QMSR). This new rule, which officially takes effect on February 2, 2026, directly incorporates ISO 13485 by reference. This is a huge move toward a more unified global approach. You can dig into the specifics of this QMSR transition to see how it might affect your operations.
Comparing FDA QSR, QMSR, and ISO 13485
To really grasp what this shift means, it helps to see the regulations side-by-side. The following table breaks down the key differences and the new alignment between the FDA's old rules, the new harmonized rules, and the global standard.
| Area of Focus | FDA QSR (Legacy) | ISO 13485:2016 (Global Standard) | FDA QMSR (Effective 2026) |
|---|---|---|---|
| Core Philosophy | Prescriptive, with specific requirements for procedures and records. | Process-based and risk-focused, emphasizing integration of risk management throughout the product lifecycle. | Harmonized with ISO 13485, adopting its process-based and risk-centric approach. |
| Risk Management | Required but less explicitly integrated throughout the entire QMS. | A central, foundational requirement woven into nearly every clause of the standard. | Directly incorporates ISO 13485's comprehensive risk management requirements. |
| Terminology | Uses specific FDA terms like "management with executive responsibility" and "design history file (DHF)." | Uses globally recognized terms like "top management" and "design and development files." | Adopts ISO 13485 terminology, with some specific FDA clarifications added. |
| Flexibility | More rigid and checklist-oriented. | More flexible, allowing organizations to tailor processes to their specific devices and risks. | Inherits the flexibility of ISO 13485, focusing on effectiveness rather than just procedural compliance. |
| Global Alignment | U.S.-specific, often requiring separate documentation for international markets. | The de facto global standard, accepted by most major regulatory authorities worldwide. | Designed for global harmonization, allowing one QMS to meet requirements for the U.S. and other major markets. |
This harmonization simplifies life for manufacturers immensely. Now, you can build one primary QMS that satisfies the world's largest medical device markets.
What Harmonization and the New FDA QMSR Really Mean
This move to the QMSR is far more than a cosmetic update. It signals a fundamental shift in how the FDA views quality, aligning its expectations with the global consensus.
So, what does this actually mean for your team on the ground?
- Risk is Everything: The QMSR, by adopting ISO 13485, puts a massive emphasis on a risk-based approach. You can no longer treat risk management as a siloed activity; it has to be part of the DNA of your entire QMS.
- Focus on the Process: The new rule steps away from the old prescriptive, "did you check the box?" style. It pushes for a more holistic view where you have to demonstrate that your processes are effective and interconnected.
- Speaking the Same Language: You'll see terms from ISO 13485, like "top management," become part of the official FDA vocabulary. This isn't just semantics—it reinforces the expectation that leadership must be deeply involved in the quality system.
This alignment is a huge win for the industry. It means companies can finally build one powerful, efficient quality management system that works for multiple regulatory bodies. That translates to less duplicated effort and a clearer path to getting products on the global market.
If your organization is already certified to ISO 13485, this transition should be fairly straightforward. But for companies that have only ever built their systems around the old QSR, it's time for a serious gap analysis to find where your system falls short of ISO 13485's requirements. As you begin that process, it’s also a good idea to understand the costs associated with ISO 13485 certification to build it into your budget.
A Quick Note on the EU MDR
While the FDA’s new rule is big news, don't forget about other major markets. The European Union's Medical Device Regulation (MDR) also requires a QMS that complies with ISO 13485.
The MDR, however, piles on its own unique requirements, especially around clinical evidence, post-market surveillance, and supply chain traceability. If you plan to sell in Europe, these extra demands must be woven directly into your quality system. The real key to a successful global strategy is understanding how all these standards and regulations interconnect.
Understanding the Core Processes of Your QMS
A medical device quality management system is so much more than a binder full of rules collecting dust on a shelf. It's the living, breathing operational engine that drives your company toward creating safe, effective products. To really get it, you need to look at its core processes as interconnected gears in a precision machine—each one depending on the others to keep things moving smoothly.
Think of it like building a skyscraper. You have architects, structural engineers, safety inspectors, and supply managers all working together. If one team drops the ball, the integrity of the entire building is at risk. Your QMS works on the exact same principle of shared responsibility and seamless integration.
This diagram shows how the key global standards governing these processes fit together, especially with the FDA's recent shift toward harmonization.
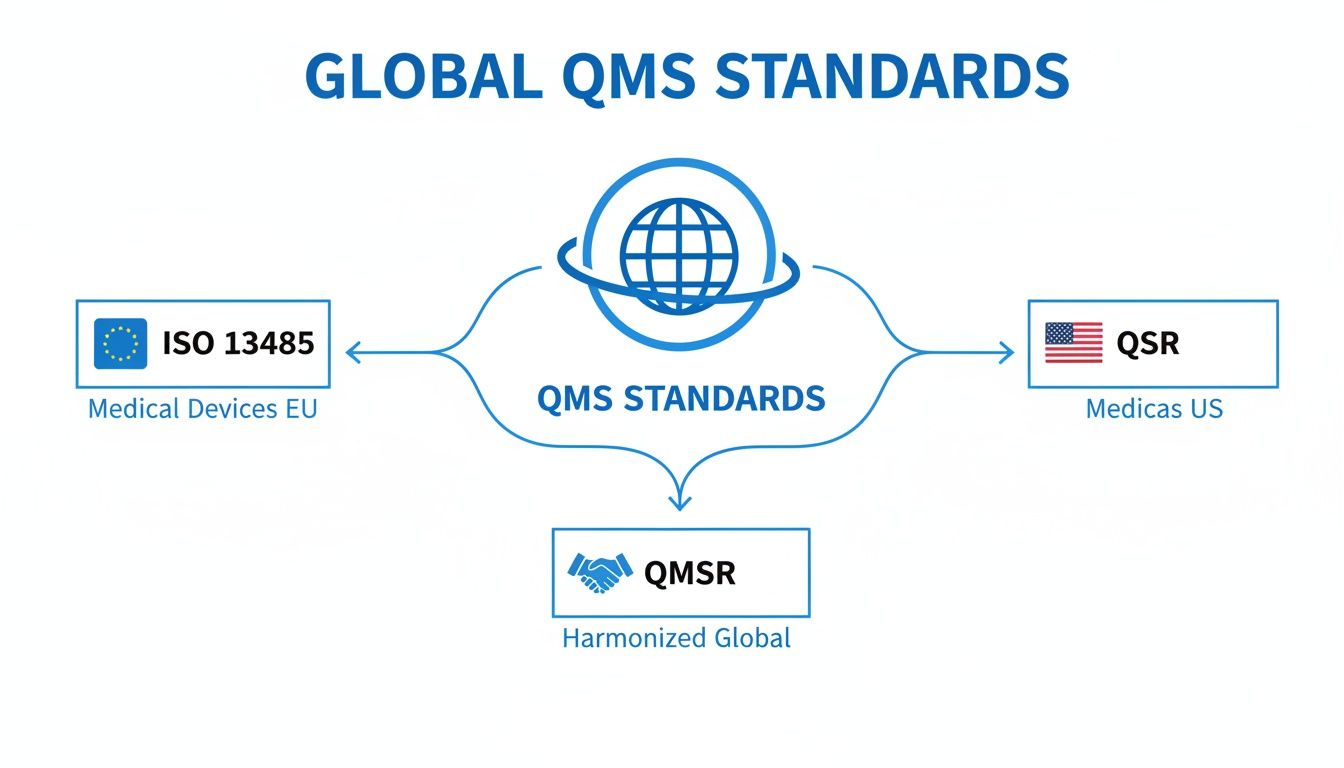
As you can see, the new QMSR essentially builds a bridge, officially linking the U.S. regulatory framework with the global ISO 13485 standard to create a more unified compliance landscape for everyone.
Design Controls: The Architectural Blueprint
You can't just inspect quality into a device at the end of the production line. It has to be engineered in from the very first napkin sketch. That’s the whole point of design controls. They are the formal, documented procedures that rein in the chaos of development, making sure you create a device that actually meets user needs and is fundamentally safe.
Think of design controls as the architectural blueprint for a house. An architect doesn't just tell the builders to "make it look nice." They create detailed plans specifying everything from the foundation’s depth to the electrical wiring. This blueprint guarantees the final structure is sound, functional, and up to code. Design controls do the same for your medical device, ensuring it's built on a solid foundation of well-defined requirements.
The process breaks down into a few key stages:
- Design Inputs: This is where you nail down what the device must do. It's all based on user needs, regulatory standards, and its intended use.
- Design Outputs: These are the "recipes" for building the device—think drawings, material specifications, and manufacturing instructions.
- Design Verification: Here, you ask, "Did we design the device right?" This step is about confirming that your outputs (the recipes) actually meet your inputs (the requirements). It’s all about testing and inspection.
- Design Validation: Now you ask, "Did we design the right device?" This is the big one. It confirms the finished product truly meets the user’s needs, which is often proven through clinical trials or usability studies.
Without this methodical approach, development is just a free-for-all, leading to expensive redesigns and, far worse, unsafe products hitting the market.
Risk Management: Your Early Warning System
While design controls provide the blueprint, risk management is your QMS's built-in early warning system. Governed by the global standard ISO 14971, this isn't a one-and-done activity. It’s a continuous process of identifying, evaluating, and mitigating potential hazards associated with your device throughout its entire lifecycle.
Imagine you're the captain of a ship. You wouldn't just wait for a storm to hit and then react. You'd use radar, weather forecasts, and navigation charts to anticipate dangers and steer clear of them. Risk management is your QMS's radar, constantly scanning the horizon for trouble.
A robust risk management process doesn't just generate a list of what could go wrong. It forces you to actively build safety measures into the device's design, manufacturing processes, and even its labeling to either eliminate hazards or reduce them to an acceptable level.
For example, a risk analysis for a battery-powered infusion pump might identify the potential for sudden power failure. Your risk controls would then be to design in a backup battery and a low-power alarm. This proactive mindset is at the very heart of modern regulatory philosophy.
CAPA: The System’s Immune Response
No matter how perfectly you plan, things will go wrong. That’s where Corrective and Preventive Action (CAPA) comes into play. CAPA is your QMS's immune response—a formal system for reacting to problems, digging deep to find their root causes, and implementing changes to ensure they never happen again.
A corrective action fixes a problem that already exists, like recalling a faulty batch of devices. A preventive action, on the other hand, addresses a potential problem before it even occurs, like updating a manufacturing process because data analysis suggests a future risk. The goal is to learn from your mistakes and make the entire system stronger.
Companies without a good CAPA system get stuck in a "firefighting" loop, solving the same issues over and over without ever addressing the underlying cause. A proper CAPA investigation is like being a detective—you have to keep asking "why" until you uncover the true root cause, which is the only way to find a permanent solution.
Document and Supplier Control: The Foundational Pillars
Finally, none of this works without the structural integrity provided by document control and supplier management. These are the unsung heroes of the QMS.
Document Control is the systematic process that ensures everyone is always working from the latest approved version of a procedure, specification, or form. Without it, you have chaos. Engineers might use outdated drawings, or assembly technicians could follow obsolete instructions, leading to disastrous consequences.
Supplier Control makes sure the components and services you buy from other companies meet your quality standards. Your device is only as good as its weakest part. This process involves thoroughly vetting suppliers, setting up clear quality agreements, and continuously monitoring their performance. It's about making sure your partners don't unknowingly introduce risks into your product. Together, these two processes form the reliable, traceable framework that your entire QMS is built upon.
Building Your Audit-Ready Evidence Trail
A top-notch medical device QMS is more than a stack of well-written procedures. It's a living, breathing system proven by a clear and undeniable trail of evidence. When an auditor walks through your door, they aren’t just interested in what your SOPs say you should do. They want to see cold, hard proof that you actually did it.
Think of it like building a case file for a meticulous detective. Every single claim you make about your quality processes, from design to production, must be backed up by interconnected, easily accessible records. This evidence weaves together a cohesive story of your device's entire lifecycle, leaving no room for an auditor to second-guess your compliance.
Your goal is to present a narrative so clear that an auditor can follow the breadcrumbs from an initial user need all the way to a finished, validated product without ever hitting a dead end or a confusing gap.
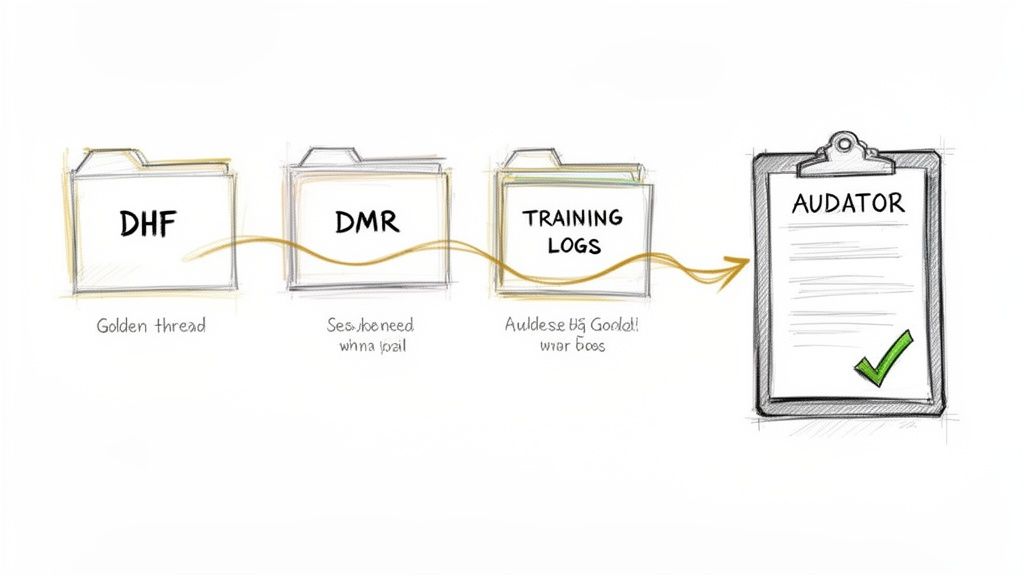
The Key Files in Your Evidence Locker
Certain documents are the foundational pillars of your audit-ready evidence trail. These aren't just administrative files; they are the comprehensive records that prove your processes were followed correctly at every critical turn.
Here are three of the most important record collections an auditor will want to see:
- Design History File (DHF): This is the complete biography of your device's design. It contains every record demonstrating the design was developed according to your approved plan, from user needs and design inputs to the final verification and validation reports. It tells the why and how behind every design decision.
- Device Master Record (DMR): If the DHF is the story of how you designed the device, the DMR is the official recipe for how to build it perfectly every single time. It contains all the drawings, material specifications, and manufacturing procedures needed to produce a consistent and safe product.
- Training Records: These files are your proof that the people designing, building, and testing your device were actually qualified for the job. They're critical for showing you have a competent team executing your quality system.
An auditor should be able to pick any of these files and see a clear, logical progression. A DHF without a final validation report is an unfinished story; a DMR with an outdated component specification is a recipe for disaster.
Traceability: The Unbreakable Golden Thread
The real test of your evidence trail is traceability. This is the "golden thread" that connects every single dot in your quality system. It’s the ability to show the direct, unbroken link between your initial customer requirements, your design inputs, the resulting design outputs, and the verification and validation activities that followed.
For instance, an auditor might point to a specific feature on your device and ask, "Prove to me this was tested properly." With strong traceability, you can instantly pull up the verification report confirming the feature met its specification, which in turn links directly back to the original design input that demanded it.
This interconnectedness is what separates a world-class medical device quality management system from a chaotic jumble of documents. A detailed traceability matrix is often the centerpiece of this evidence, providing a clear roadmap of these crucial connections.
Unfortunately, many companies still try to manage this intricate web using scattered spreadsheets, shared drives, and paper binders. This approach is notoriously fragile. A single broken link in a spreadsheet or a misplaced document can bring an audit to a screeching halt, leading to costly findings and delays. For teams getting ready for an inspection, using a solid audit readiness checklist can help spot these weak points before they become major headaches.
Ultimately, a seamless, low-stress audit depends on creating an evidence trail that is not just complete, but also transparent and incredibly easy for an outsider to navigate.
How AI Can Take the Pain Out of QMS Compliance
Anyone who has prepared for a QMS audit knows the drill. You spend weeks, sometimes months, digging through a mountain of documentation—SOPs, validation reports, risk files—trying to connect every piece of evidence to the right regulatory clause. It's a painful, manual slog that’s not only a massive time-sink but also ripe for human error.
Now, imagine a different approach. Instead of your team spending hundreds of hours hunting for documents, an AI assistant does the heavy lifting, reading and understanding your entire QMS library in a matter of minutes. These tools can automatically map your existing evidence directly to the specific clauses in standards like ISO 13485 or the FDA’s requirements.
This isn’t about replacing quality professionals. Far from it. It's about giving them a powerful new tool so they can stop being document detectives and start focusing on what they do best: improving quality and ensuring safety.
From Manual Drudgery to Strategic Focus
The real magic of AI-powered compliance tools is how they can turn weeks of grueling prep work into just a few hours of focused, strategic review. By automating that initial, tedious phase of finding evidence, your team gets to jump straight to the important part: analyzing the findings and understanding your true compliance health.
Here’s how they make it happen:
- Evidence-Linked Reporting: The AI produces reports where every claim is directly tied to the source evidence, often citing the exact page number. No more guesswork.
- Deep Links to Source Documents: With a single click, you can go from a gap in your report straight to the precise paragraph in the original PDF. This makes verification almost instantaneous.
- Automatic Gap Spotting: The system is smart enough to flag areas where your evidence is thin or completely missing, creating a clear, prioritized to-do list for your team.
This completely changes the dynamic of audit preparation. It shifts the entire process from a reactive, last-minute scramble to a proactive, continuous cycle of analysis and improvement. Suddenly, being "audit-ready" all the time doesn't seem like a fantasy.
The industry is already moving in this direction. Since 2022, there's been a 40% jump in firms adopting cloud-based QMS solutions. These newer platforms can integrate with other business systems for live monitoring and have been shown to cut compliance-related costs by 25-35%. For a quality manager, this means a SaaS tool can analyze all your documents against ISO 13485 and shrink audit prep time from months down to days. You can find more details on this trend over at Market Report Analytics.
Putting Powerful Compliance Tools Within Reach
These AI systems are built to bring sophisticated compliance management to companies of any size, not just the giants. The experience is often centered around a single, shared workspace where the whole team can manage the gap assessment process.
This screenshot gives you a feel for it. A user simply uploads their QMS documents and chooses the standard they want to assess against.
The platform then lays everything out in a clear, organized way. Teams can manage their evidence, pinpoint gaps, and work together on fixing them, all in one place. By taking the most labor-intensive tasks off your team's plate, these tools help you build a stronger, more resilient medical device quality management system. To learn more about this approach, take a look at our guide on the modern gap assessment process.
Frequently Asked Questions About Your Medical Device QMS
It’s one thing to understand the theory behind a medical device quality management system, but it's another to live with it day-to-day. Let's tackle some of the practical questions that always come up when you're in the trenches building and running a QMS.
How Long Does It Take to Implement a QMS?
There's no magic number here. The timeline really depends on the size of your company, how complex your device is, and frankly, the state of your current documentation (or lack thereof).
A small startup working on a simple Class I device might pull it off in 6 to 12 months if they’re focused. On the other hand, a larger, established company with a high-risk device or multiple product families should probably budget for 12 to 18 months, maybe even more.
The best advice? Don’t treat it like a sprint to the finish line. Think of it as pouring the foundation for your company's future. A phased approach always works best, and using dedicated QMS software can give you a huge head start with built-in templates and proven workflows.
What’s the Difference Between a QMS and an eQMS?
This question comes up all the time, and it's a good one because the terms get thrown around interchangeably. They are definitely not the same thing.
- A QMS (Quality Management System) is the system itself—all the policies, procedures, and processes that define how you operate. It's the "what" and "why" behind your quality efforts.
- An eQMS (electronic Quality Management System) is the software platform you use to run that system. This is the tool that automates document control, tracks training, manages CAPAs, and organizes your design history files.
Here’s an analogy: Your QMS is the playbook for a championship team, detailing every strategy and role. The eQMS is the digital communication system they use on the field—headsets, tablets, and real-time analytics—to execute that playbook flawlessly. An eQMS takes your quality system from a bunch of binders on a shelf to a living, connected ecosystem.
An eQMS turns a static collection of documents into a dynamic, single source of truth. When an auditor asks for evidence, it’s all right there—connected, traceable, and under control.
Is ISO 13485 Certification Mandatory?
This is where things get interesting. Legally speaking, ISO 13485 certification isn't a hard-and-fast requirement in every single country on the planet. But in practice? It absolutely is if you want to sell in any major market.
For instance, you can't get very far in places like Canada or the European Union without it; it's a standard regulatory hurdle. In the U.S., the FDA has its own rules, the Quality Management System Regulation (QMSR), but they've now aligned them so closely with ISO 13485 that the standard is the clearest path forward.
So, while you might find a corner of the world where it's not technically "mandatory," getting ISO 13485 certified is the most efficient way to prove your medical device quality management system is up to snuff for the FDA and other global regulators. It's become the universal passport for market access.
Ready to transform your audit preparation from a manual marathon into a strategic sprint? AI Gap Analysis uses artificial intelligence to read your entire QMS library, map evidence to regulatory clauses, and pinpoint gaps in minutes, not months. Stop hunting for documents and start focusing on quality.