February 15, 2026
regulatory compliance for medical devicesmedical device complianceISO 13485quality management systemFDA compliance
Mastering Regulatory Compliance for Medical Devices
Navigate regulatory compliance for medical devices with this guide. Learn to master QMS, risk management, and audit readiness in the complex global market.
19 min readAI Gap Analysis

Regulatory compliance for medical devices isn't just about paperwork; it's the disciplined process of proving your device meets every legal, safety, and quality standard set by government authorities before it can ever be sold. This means embedding strict regulations, like the FDA's Quality Management System Regulation (QMSR) in the U.S. and the EU's Medical Device Regulation (MDR), into the very DNA of your product development. Success comes from a proactive, systematic commitment to quality and risk management right from the start.
Navigating the High Stakes World of Device Compliance
Think of bringing a medical device to market as navigating a complex global shipping network. Every port—representing a different country or region—has its own set of non-negotiable customs rules, documentation requirements, and inspection protocols. A mistake in your paperwork for one port can get your entire shipment impounded, causing costly delays and damaging your reputation.
The medical device world works much the same way, but the stakes are exponentially higher. You’re navigating a maze of international regulations from bodies like the U.S. Food and Drug Administration (FDA), the European Union with its stringent MDR framework, and other global authorities. Each has its own distinct and demanding requirements. Failing to meet these standards isn’t just a logistical headache; it can have severe, business-altering consequences.
The Real Cost of Non-Compliance
Ignoring or mishandling these obligations isn't a minor slip-up. It's a foundational failure that can cripple a company's ability to operate and put patients at risk.
- Market Denial or Withdrawal: The most immediate threat is being blocked from selling your product. Regulators can refuse market access entirely or force a recall of devices already in use, erasing revenue streams overnight.
- Legal and Financial Penalties: The fines for non-compliance can be staggering. Something as seemingly simple as failing to report adverse events can trigger severe penalties that easily run into the millions.
- Reputational Damage: A public recall or a regulatory warning letter devastates the trust you've built with patients, healthcare providers, and investors. Rebuilding that confidence is a long, expensive, and sometimes impossible process.
Navigating the regulatory landscape isn't about checking boxes. It’s about building a culture of quality and safety that protects patients and proves to regulators that your device is both effective and safe for its intended use.
This guide is your strategic roadmap. We'll break down the essential components of regulatory compliance for medical devices, turning what feels like a daunting obligation into a clear, manageable process.
To get started, it's helpful to see how the major players fit together. Each regulatory body has a different focus, and understanding their roles is the first step toward building a global compliance strategy.
Key Global Regulatory Frameworks at a Glance
| Regulatory Body / Framework | Geographic Region | Primary Focus |
|---|---|---|
| U.S. Food and Drug Administration (FDA) | United States | Governs pre-market approval (PMA), 510(k) clearance, and the Quality Management System Regulation (QMSR). |
| European Union (MDR & IVDR) | European Union/EEA | Enforces comprehensive rules for medical devices (MDR) and in-vitro diagnostic devices (IVDR) via Notified Bodies. |
| Health Canada | Canada | Regulates devices through the Medical Devices Regulations, requiring a Medical Device License (MDL). |
| Therapeutic Goods Administration (TGA) | Australia | Oversees device registration in the Australian Register of Therapeutic Goods (ARTG). |
| International Medical Device Regulators Forum (IMDRF) | Global | Works to harmonize regulatory requirements worldwide, influencing standards like ISO 13485. |
Understanding these core frameworks is crucial. By mastering the principles behind quality management, clinical evidence, and post-market surveillance, you can build a system that not only satisfies auditors but also creates a powerful competitive advantage. A strong compliance posture accelerates market access, builds lasting trust, and ultimately ensures your innovations can safely reach the patients who need them.
Keeping Pace with Constant Regulatory Changes
The rulebook for medical devices isn’t written in stone. It’s a living document, constantly being edited and expanded. For regulatory affairs teams, this creates a relentless challenge. Staying compliant means more than just understanding today's rules—it requires a sharp eye on what's coming next.
Think of it like being the captain of a ship on a long voyage. You’re not just navigating the current weather; your charts are being updated in real-time with new currents, newly discovered hazards, and changing port entry requirements. This is the reality for medical device manufacturers, where the regulatory landscape is in a state of perpetual motion.
The Ever-Shifting Global Frameworks
Major regulatory shifts are happening all at once across key markets, and each one demands significant attention and resources. The European Union's move to the much tougher Medical Device Regulation (MDR) and In-Vitro Diagnostic Regulation (IVDR) is a perfect example. This wasn't a minor update; it was a fundamental overhaul that reset expectations for clinical evidence, post-market surveillance, and traceability.
At the same time, post-Brexit, the UK is establishing its own framework with the UKCA mark, creating a separate set of rules for market access in Great Britain. Across the pond, the U.S. FDA is harmonizing its long-standing Quality System Regulation (QSR) with the international standard ISO 13485, creating the new Quality Management System Regulation (QMSR). Each of these changes, while aimed at improving safety, adds another layer of complexity.
And these aren't isolated events. The pace is only accelerating. Between 2020 and 2024, the MedTech sector had to navigate over 15 landmark regulations, more than 60 major guidelines, around 100 technical amendments, and at least 20 harmonization efforts globally. This barrage hits regulatory professionals the hardest, with the EU's MDR/IVDR transitions alone flagging over 80% of legacy devices for re-certification under tighter deadlines. You can learn more about how regulatory compliance for medical devices is evolving in this detailed guide from RegDesk.
The core challenge is no longer just achieving compliance, but maintaining it. The finish line is constantly moving, and a static, 'set-it-and-forget-it' approach is a direct path to non-compliance.
The Real-World Impact on Your Operations
These high-level changes create very real, ground-level pressures on your organization. The global push toward greater transparency and patient safety is forcing manufacturers to rethink their entire product lifecycle management from the ground up.
Key operational impacts include:
- Re-certification of Legacy Devices: Products that have been on the market for years under older rules (like the MDD in Europe) don't get a free pass. They have to go through a rigorous re-assessment against the new, stricter criteria of the MDR. Our MDR timeline calculator can be a big help in planning for this.
- Stricter Traceability Demands: The rollout of Unique Device Identification (UDI) systems means every single device must be trackable from the factory floor to the patient. This requires rock-solid systems to manage and report data accurately.
- Increased Clinical Evidence Requirements: Regulators now demand far more comprehensive clinical data to back up safety and performance claims—not just at launch, but throughout the device's entire lifecycle through active post-market surveillance.
Trying to track all these updates manually with spreadsheets and email alerts is becoming an impossible task. The sheer volume and speed of change are just too much. A single missed update to a harmonized standard or a new guidance document can put your market access at risk. This environment makes a strong case for moving away from manual methods and toward more systematic, tech-driven solutions that can keep up with the constant evolution of regulatory compliance for medical devices.
Building Your Foundation on Quality and Risk Management
If getting a medical device to market is like constructing a building, your Quality Management System (QMS) is the foundation. It’s not just a pile of documents or a box-ticking exercise for auditors. A truly effective QMS is the very blueprint for how your company operates, ensuring safety and quality are embedded in your device from day one.
Think of it as your company's central nervous system. It connects everything—from R&D and supplier vetting to manufacturing and post-market surveillance—into a single, traceable system. This ensures every decision is documented, every process is repeatable, and every component is controlled.
ISO 13485: The Global Standard for Medical Device Quality
At the core of nearly every medical device QMS is ISO 13485. This isn't just a helpful guideline; it's the globally recognized standard for quality in our industry. Regulators worldwide, including the FDA with its updated Quality Management System Regulation (QMSR), view it as the definitive benchmark.
Adopting ISO 13485 means committing to a structured, lifecycle-wide approach. The whole point is to consistently design, build, and support devices that not only work as intended but also meet the strictest regulatory demands.
Key pillars mandated by ISO 13485 include:
- Management Responsibility: Leadership isn't passive. The standard requires top-down commitment, with clearly defined roles and responsibilities for maintaining quality.
- Resource Management: You must have the right people, tools, and environment to get the job done properly—and be able to prove it.
- Product Realization: This is the big one. It covers the entire journey from design and development planning through purchasing, production, and service.
- Measurement and Analysis: You have to monitor your processes, conduct internal audits, and manage nonconforming products to fuel continuous improvement.
For a deeper dive into this foundational standard, consider exploring our comprehensive guide on building a medical device quality management system.
ISO 14971: The Inseparable Partner to Your QMS
A QMS gives you the "how," but it needs a partner to address the "what if." That's where risk management, governed by ISO 14971, comes in. Quality and risk are two sides of the same coin; you simply can't manage one without the other.
ISO 14971 requires a systematic process for identifying, evaluating, and controlling risk across your device's entire lifecycle. This isn't a one-and-done task you complete before launch. It's a living process that continues long after your device is in the hands of users.
A QMS without integrated risk management is like a car with a great engine but no brakes. It may move forward, but it lacks the critical mechanism to navigate hazards and ensure a safe journey for the user.
When an auditor shows up, they will look for evidence that risk management is deeply integrated into your daily operations, not just sitting in a folder. They'll expect to see a comprehensive risk management file that clearly shows how you've:
- Identified potential hazards associated with your device.
- Estimated and evaluated the risks these hazards pose to patients and users.
- Implemented control measures to drive those risks down to an acceptable level.
- Monitored the effectiveness of those controls over time.
For instance, a risk file for an infusion pump would identify a software bug causing an overdose as a critical hazard. The file would then detail the specific controls—like redundant code reviews, automated testing protocols, and built-in alarms—used to mitigate that risk. This entire thought process has to be woven directly into the fabric of your QMS.
Ultimately, getting your foundation right with quality and risk management does more than just please regulators. It creates a culture of accountability, prevents expensive post-market problems, and builds the trust you need with the clinicians and patients who rely on your device.
Putting the Essential Pillars of Your Strategy into Practice
With a solid Quality Management System (QMS) as your foundation, the next step is to build out the operational pillars that support your device through its entire lifecycle. These aren't just siloed activities; they are deeply interconnected systems that work together to prove your device is safe, effective, and meticulously documented.
Think of it this way: your QMS is the facility, but these pillars are the critical departments inside. You have the R&D lab (Clinical Evaluation), the 24/7 security and monitoring team (Post-Market Surveillance), and the central archives (Technical Documentation). A failure in one department puts the entire operation at risk.
Proving Your Device Works with Clinical Evaluation
Before you can claim your device helps a single patient, you have to prove it. That's the entire point of the Clinical Evaluation Report (CER). This isn't just about running one study; it's a continuous process of gathering, appraising, and analyzing clinical data to confirm your device performs as intended without introducing unacceptable risks.
Imagine you're building a legal case. The CER is your complete evidence binder, presenting a logical and unbiased argument that is backed by credible, verifiable data. You can be sure that auditors will scrutinize this report to see if your claims truly hold up.
A strong clinical evaluation process always includes:
- Gathering Clinical Data: This pulls from clinical investigations of your device, scientific literature on similar devices, and any clinical experience from those equivalent products.
- Appraising the Data: You have to critically assess the quality and relevance of every piece of data. Does it genuinely support your claims about safety and performance?
- Analyzing and Concluding: The final step involves analyzing all that appraised data to determine if you have enough evidence to confirm compliance with all regulatory requirements.
And remember, this isn't a "one-and-done" task. The CER must be actively updated throughout your device's life on the market, incorporating new findings from your post-market activities.
Listening to the Real World with Post-Market Surveillance
Once your device hits the market, your work is really just beginning. Post-Market Surveillance (PMS) is your ongoing "listening system," designed to systematically collect and review real-world experience with your device. It’s how you proactively spot emerging risks and confirm your product continues to perform as expected out in the wild.
Think of it as the crucial feedback loop between your company and the clinicians and patients using your device. Without it, you’re flying blind, completely unaware of potential issues until they escalate into major problems. A robust PMS plan is simply non-negotiable for modern regulatory compliance for medical devices.
A proactive Post-Market Surveillance plan is the difference between identifying a potential issue from a handful of user reports and reacting to a full-blown crisis that requires a recall. It’s your early warning system.
The growing emphasis on this area is clear from market trends. The global medical device regulatory affairs market was valued at USD 6.7 billion in 2024 and is projected to hit USD 11.66 billion by 2030. This growth is fueled by stricter oversight and new technologies, with systems like the EU's EUDAMED intensifying PMS traceability. Falling short here can lead to certification delays of up to 18 months. You can explore more about these market trends on Grand View Research.
Maintaining Your Master Evidence File
Finally, all this proof needs a place to live. Your technical documentation, often called the Technical File or Design Dossier, is the master repository that substantiates every single claim about your device. It's the comprehensive file that proves to regulators you’ve done your homework.
This file tells the complete story of your device, from its initial concept to its final design, manufacturing process, risk analysis, and post-market monitoring. It connects everything, providing a clear and traceable path that an auditor can follow to verify every claim you make. Neglecting this file is one of the fastest ways to fail an audit.
How to Actually Be Ready When the Auditors Arrive
Knowing the regulations for medical devices is one thing. Proving you follow them when an auditor is sitting across the table? That’s an entirely different challenge. When they ask for specific evidence, the pressure mounts, and the clock starts ticking.
For too many teams, this moment triggers a frantic scramble. They dive into a maze of shared drives, email chains, and disconnected documents, desperately trying to stitch together a coherent story. This old-school approach isn't just slow—it’s a recipe for human error, missed evidence, and half-answers that can make an auditor lose confidence in a hurry.
The Old Way vs. The New Way
Trying to gather evidence manually is like being asked to find a specific sentence in a library where none of the books are organized. You know the information is somewhere, but finding it is a slow, painful process. You might grab the wrong book, misinterpret a page, or completely miss the connection between two different documents.
This outdated process is riddled with risks:
- Incomplete Evidence: Key documents get overlooked, leading to non-conformities that were entirely preventable.
- Wasted Time: Quality and regulatory experts spend weeks just preparing for an audit instead of focusing on proactive improvements.
- Inconsistent Answers: Different people on your team might pull conflicting information, presenting a disorganized and unprofessional front.
Now, imagine a different approach. Instead of a manual library search, think of it as using a specialized search engine built to speak the language of regulations. It doesn't just look for keywords; it understands the context and intent behind the rules.
Unlocking Efficiency with AI-Powered Gap Analysis
The real breakthrough in audit preparation is the arrival of AI-powered gap analysis. These platforms are designed to consume your entire body of documentation—your QMS procedures, design history files, and risk assessments—and map it directly to the specific clauses of a standard like ISO 13485.
This screenshot shows how a modern platform can automatically map uploaded documents against compliance requirements, providing instant, evidence-linked answers.
Instead of searching by hand, the system serves up the exact evidence you need, backed by direct citations. It drastically cuts down the time it takes to prove you're in compliance.
This is especially powerful for governance, risk, and compliance (GRC) teams and consultants. They’re constantly juggling the demands of ISO 13485, ISO 9001, and new AI regulations. For them, AI gap analysis tools are a lifesaver. They can auto-parse documents against multiple frameworks, pinpoint deficiencies with deep links to the source, and let teams collaborate in real-time to create verifiable, audit-proof outputs. The result? Compliance timelines can be slashed by as much as 60%, all while keeping the expert in the loop. You can find more on this topic, including the push for greater regulatory reliance at Emergo by UL.
AI doesn't replace human judgment; it supercharges it. By taking on the tedious, repetitive work of finding evidence, these tools free up your experts to focus on what they do best: strategic analysis and fixing problems. You move from being simply prepared to being truly audit-ready.
This is a fundamental shift from reactive scrambling to proactive readiness. You can run a comprehensive gap analysis weeks or even months before the official audit, giving you a clear, actionable punch list of what needs to be fixed. It turns the audit from a high-stakes exam into a straightforward validation exercise.
By embracing this kind of automation, you build a more resilient and efficient system for managing regulatory compliance for medical devices. You can learn more about how to choose the right compliance assessment software in our detailed guide.
Your Path to Sustainable Audit Readiness
Getting a medical device through regulatory compliance isn't a one-time sprint; it's a marathon of continuous commitment to quality and patient safety. The goal is to achieve sustainable audit readiness. This means shifting your mindset away from a reactive, last-minute scramble to a proactive, evidence-first strategy.
This journey is about more than just passing an inspection. It’s about building a resilient quality system that instills confidence, protects patients, and actually helps you get to market faster.
When you embed readiness into your day-to-day operations, an audit simply becomes a moment to validate the great work you're already doing. It's no longer a frantic hunt for documents but a calm demonstration of your processes.
Charting Your Implementation Course
Getting started doesn't require a complete overhaul overnight. Instead, it's about taking deliberate, high-impact steps to build a logical and repeatable process that strengthens over time.
A practical implementation plan looks something like this:
- Centralize Your Documentation: First things first, you need to break down those information silos. Create a single source of truth where all your QMS documents, technical files, and risk assessments live. This is the bedrock of any solid compliance program.
- Conduct a Baseline Gap Assessment: You can't fix what you can't see. Before you start making changes, you have to know where your gaps are. Using modern tools to run an evidence-linked analysis against key standards like ISO 13485 gives you a clear, actionable roadmap for what to tackle first.
- Establish a Continuous Monitoring Loop: True readiness means your QMS is a living, breathing system. You need to connect your findings back to your post-market surveillance (PMS) and CAPA systems, ensuring that real-world feedback and internal audit results drive constant improvement.
This map shows the two very different paths companies can take to prepare for an audit. It starkly contrasts the old-school manual approach with a modern, automated one.
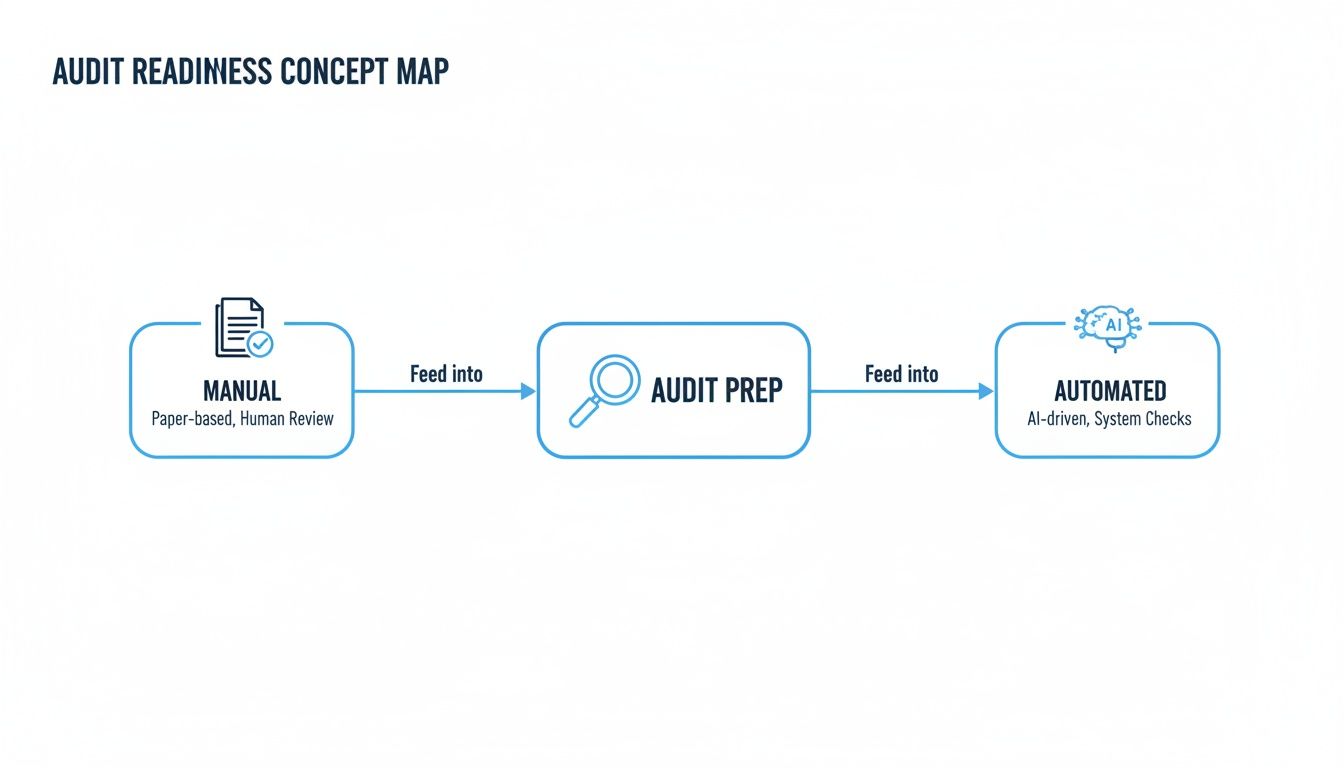
As you can see, the automated, AI-driven process is a much more direct and efficient route to getting—and staying—prepared.
By following this path, you stop just chasing compliance and start living in a state of sustainable readiness. This frees up your team to spend less time digging for documents and more time innovating and improving patient outcomes.
Ultimately, robust regulatory compliance for medical devices is your competitive edge. It’s the framework that ensures your innovations are not only groundbreaking but also fundamentally safe, building unshakable trust with regulators, healthcare providers, and the patients who depend on your technology. This dedication to quality is what secures lasting success in the market.
Frequently Asked Questions
Even with a good grasp of the fundamentals, the world of medical device compliance can leave you with some lingering questions. Let's tackle a few of the most common ones that come up.
ISO 13485 vs. FDA QSR: What's the Real Difference?
It’s easy to get tangled up when comparing ISO 13485 and the FDA's Quality System Regulation (QSR), also known as 21 CFR Part 820. The best way to think about it is that they are two different roadmaps leading to the same destination: a safe, effective product.
ISO 13485 is the globally recognized standard. Getting certified shows the world—and potential partners—that you're serious about quality. It's a voluntary standard, but it's table stakes for playing on the international stage. The FDA's QSR, however, is the law of the land in the United States. If you want to sell your device there, you have to follow it.
The good news is that they are very similar, and the FDA is moving to align its rules even more closely with ISO 13485. This new framework, called the Quality Management System Regulation (QMSR), aims to simplify things for manufacturers selling both in the U.S. and abroad.
How Does Compliance Change for SaMD?
Software as a Medical Device (SaMD) is a whole different ballgame. You can't just apply the old hardware playbook and expect it to work. Software is alive; it changes, it gets updated, and it introduces risks that a physical scalpel never could. Regulators, like the IMDRF, have created specific frameworks to handle this.
When dealing with SaMD, you have to put a much stronger emphasis on a few key areas within your QMS:
- Cybersecurity: This isn't just a "nice-to-have." Protecting patient data and ensuring your software can't be compromised is a core regulatory expectation.
- Version Control: You need an ironclad process for managing every update, patch, and new release. How do you roll it out? How do you track its performance?
- Algorithm Validation: If your SaMD uses machine learning or AI, you have to prove that your algorithm works as intended and, more importantly, that it's safe.
These aren't just extra boxes to tick; they are fundamental to proving your SaMD is safe and effective.
The single biggest compliance mistake is viewing it as a one-time event. Your QMS and technical files aren't trophies to be put on a shelf; they are living, breathing systems that must be constantly maintained to be truly audit-ready.
What Is the Biggest Compliance Mistake to Avoid?
I see it all the time: companies treat compliance like a mad dash to a finish line. They pull all-nighters to get their documentation ready for a submission or an audit, and the second it’s over, they breathe a sigh of relief and let everything collect digital dust.
That "set it and forget it" mindset is the most dangerous trap in this industry. Your Quality Management System isn't a static binder of papers; it's the operational pulse of your organization. It has to evolve with feedback from the market, with findings from your internal audits, and with every shift in the regulatory landscape. Real, sustainable audit-readiness means your evidence is always current and accurately reflects how you actually do business.
Ready to stop scrambling and achieve sustainable audit readiness? AI Gap Analysis transforms your compliance process. Upload your documents, and our AI-powered platform delivers an evidence-ready gap assessment in minutes, not weeks. Discover gaps and accelerate your path to compliance today.